
Results
Design
We have harnessed genetic engineering techniques to design the following plasmids. At the heart of our research is the plasmid pTWV228, a pivotal tool in the field of genetic engineering, and we will elucidate its details below.
pTWV228 Plasmid:
- pTWV228 is a special plasmid vector that encompasses the replication origin of pBR322, the ampicillin resistance gene from pUC118, the intergenic region IG (from M13 phage DNA), and a multi-cloning site.
- This plasmid can be cut with numerous restriction enzymes, except for the Acc I site, making it suitable for the incorporation of foreign genes.
- Furthermore, pTWV228 typically exhibits lower copy numbers compared to standard pUC-based vectors, reducing the risk of host toxicity when containing highly expressed genes.
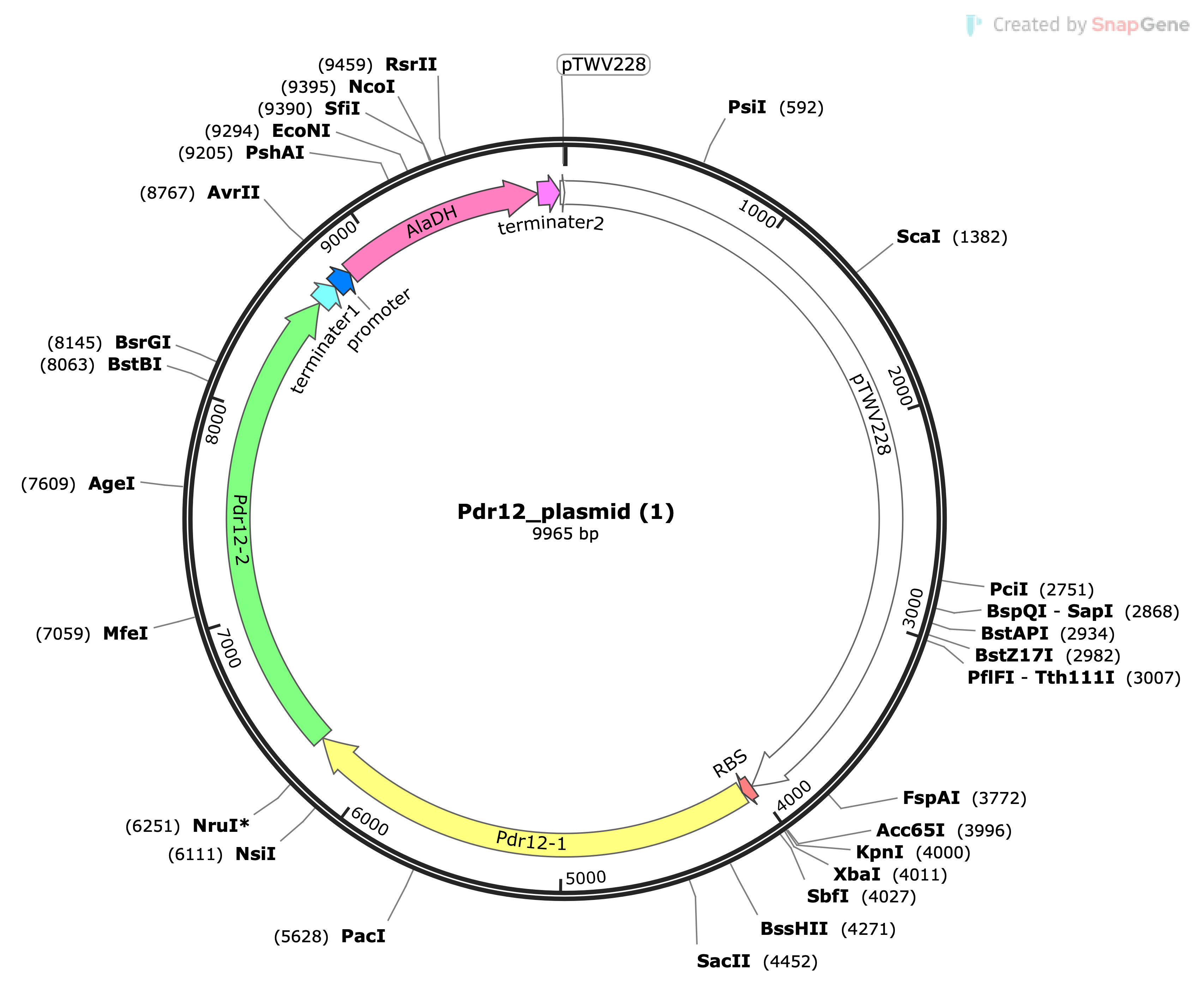
Pdr12:
- The Pdr12 gene was introduced to facilitate the active transport of pyruvic acid, a monocarboxylic acid, out of E. coli cells. This active transport regulates intracellular pyruvic acid concentrations, thereby impacting energy production and metabolic processes.
- The incorporation of the Pdr12 gene is expected to contribute to the development of novel biological functions and improvements in drug responsiveness.
AlaDH:
- The AlaDH gene was introduced to target the amino group transfer reaction from alanine to pyruvic acid. This reaction is a critical step in alanine metabolism and contributes to the regulation of intracellular balance.
- The integration of the AlaDH gene opens up possibilities for new research in alanine metabolism and the development of biotechnological applications, with potential contributions to health maintenance and optimization of production processes.
Build
During the build phase, we effectively joined all gene fragments using the Gibson Assembly method. This technique allowed us to precisely arrange specific DNA sequences to construct a recombinant plasmid.
Subsequently, the obtained recombinant plasmid was introduced into Escherichia coli cells and cultured in a medium containing ampicillin. Ampicillin was used for selective growth of bacteria harboring the plasmid (pTWV228 plasmid contains an ampicillin resistance gene). Through this cultivation process, we promoted the proliferation of genetically modified E. coli and stably maintained the plasmid.
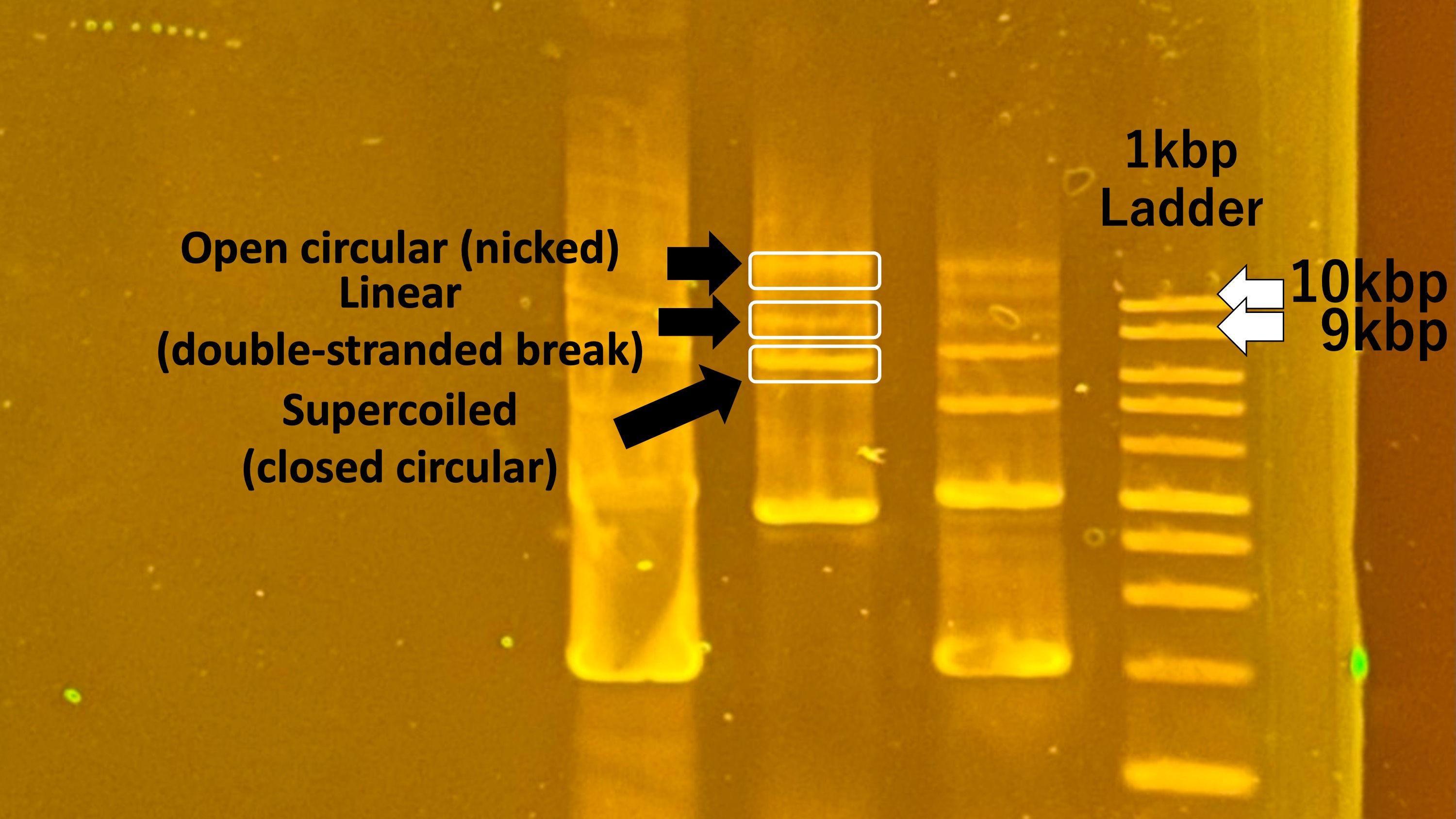
Next, we selected 3-5 colonies and cultured them in LB liquid medium using a water bath. The water bath provided an optimal temperature for cell growth and supported plasmid amplification. Following this, DNA was extracted from the cultured cells, and a mini-prep was performed. This mini-prep allowed us to confirm whether the designed recombinant plasmid was correctly introduced.
Below is an image of the DNA electrophoresis results following the mini-prep.
Test
We introduced the designed plasmid into Escherichia coli cells in which the aceF gene, a crucial gene involved in pyruvate metabolism, was knocked out. The aceF gene codes for a subunit of pyruvate phosphate dikinase (PPDK), an enzyme that catalyzes the conversion of pyruvate to acetyl-CoA. We monitored their growth until they reached an OD600 (optical density) of 2.0.
Subsequently, we cultured them in a medium containing alanine at three different concentrations (500μM, 1mM, and 2mM). Approximately every hour, we collected samples, removed E. coli cells by centrifugation, and then measured the pyruvate concentration in the extraction fluid.
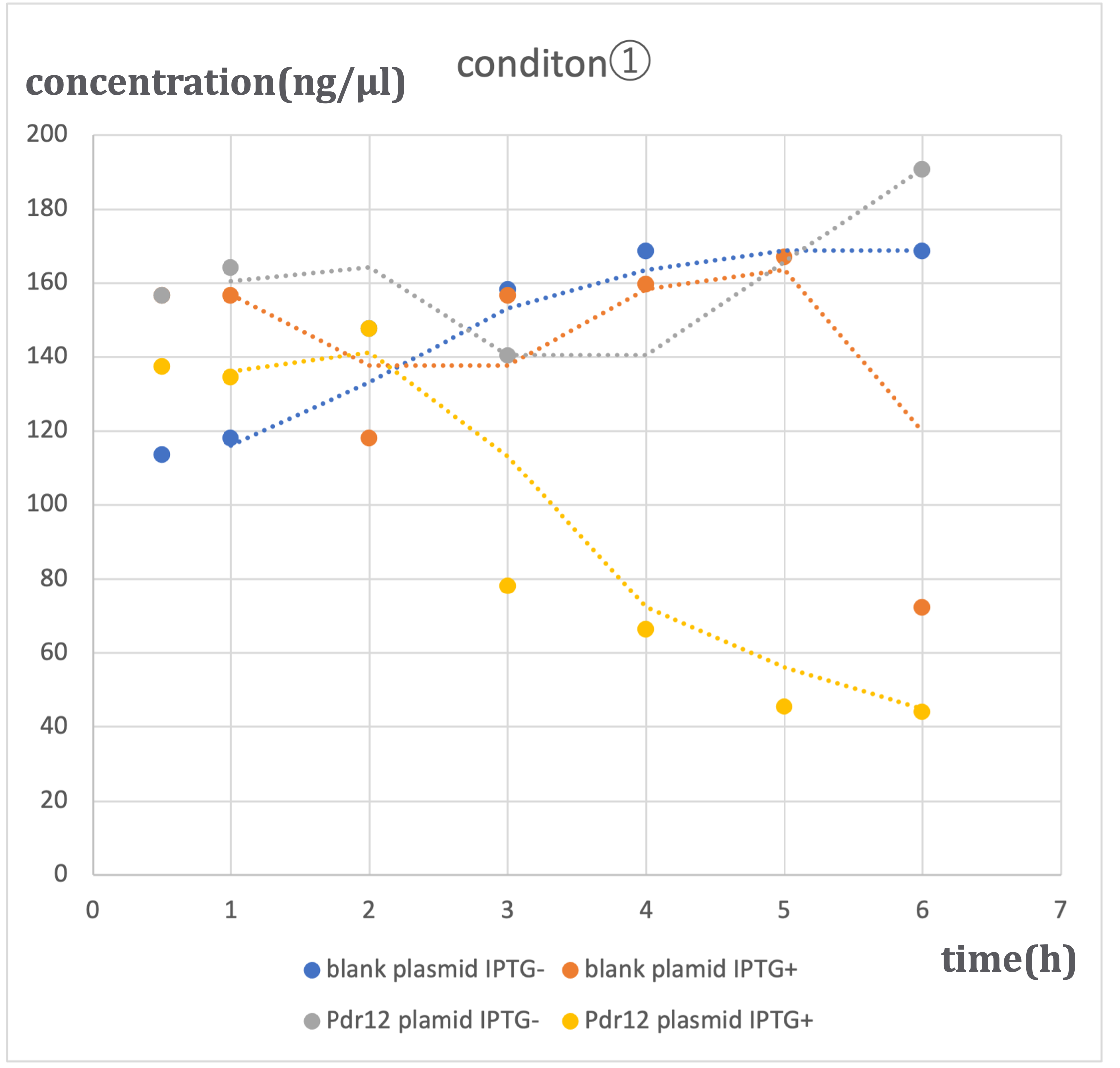
In this experiment, we conducted pyruvate concentration measurements using two types of plasmids: the designed Pdr12 plasmid and a blank plasmid (pTWV228 plasmid alone). These measurements were performed under two conditions of IPTG induction (ON and OFF), resulting in a total of four patterns. The results are as follows:
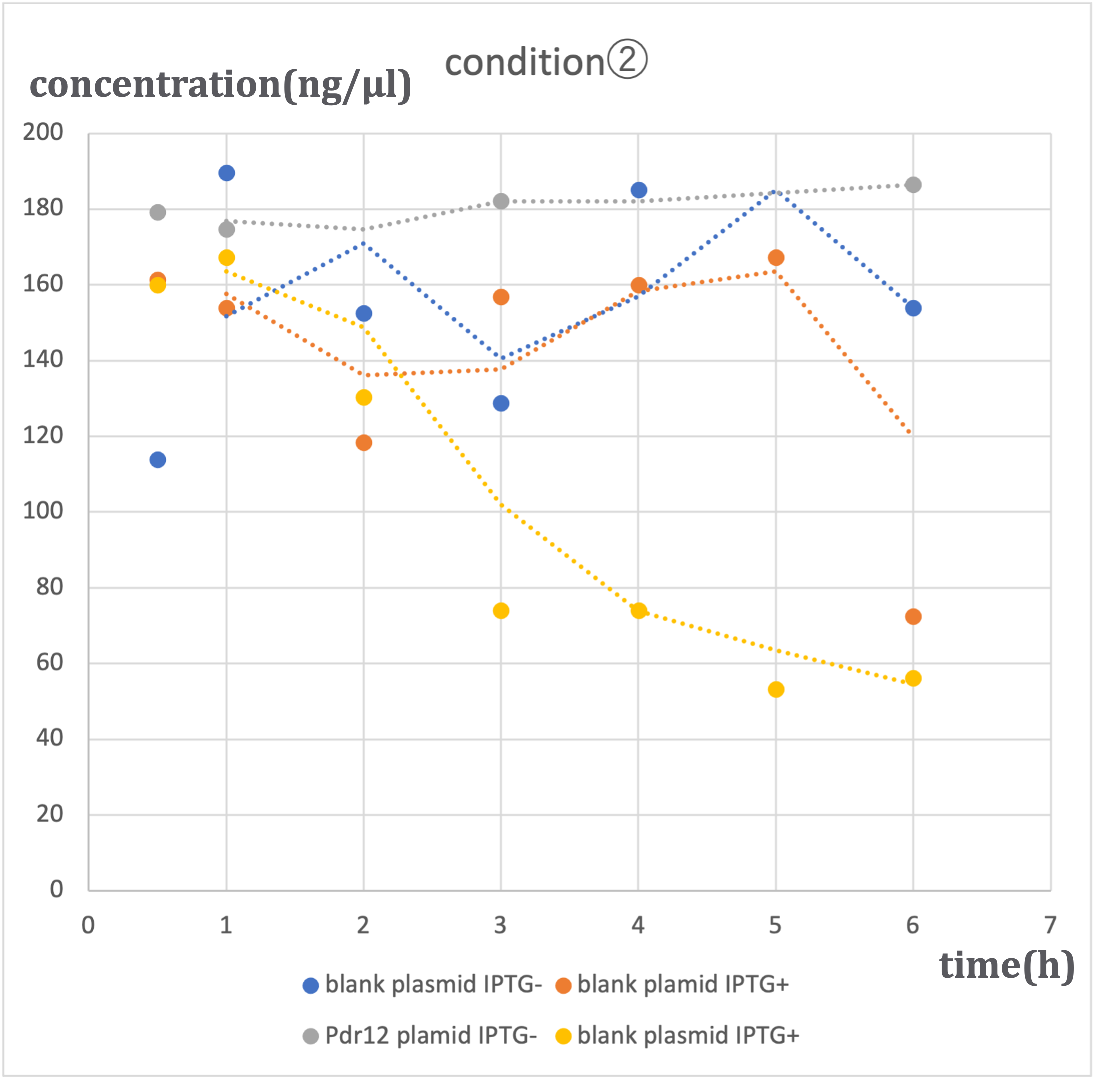
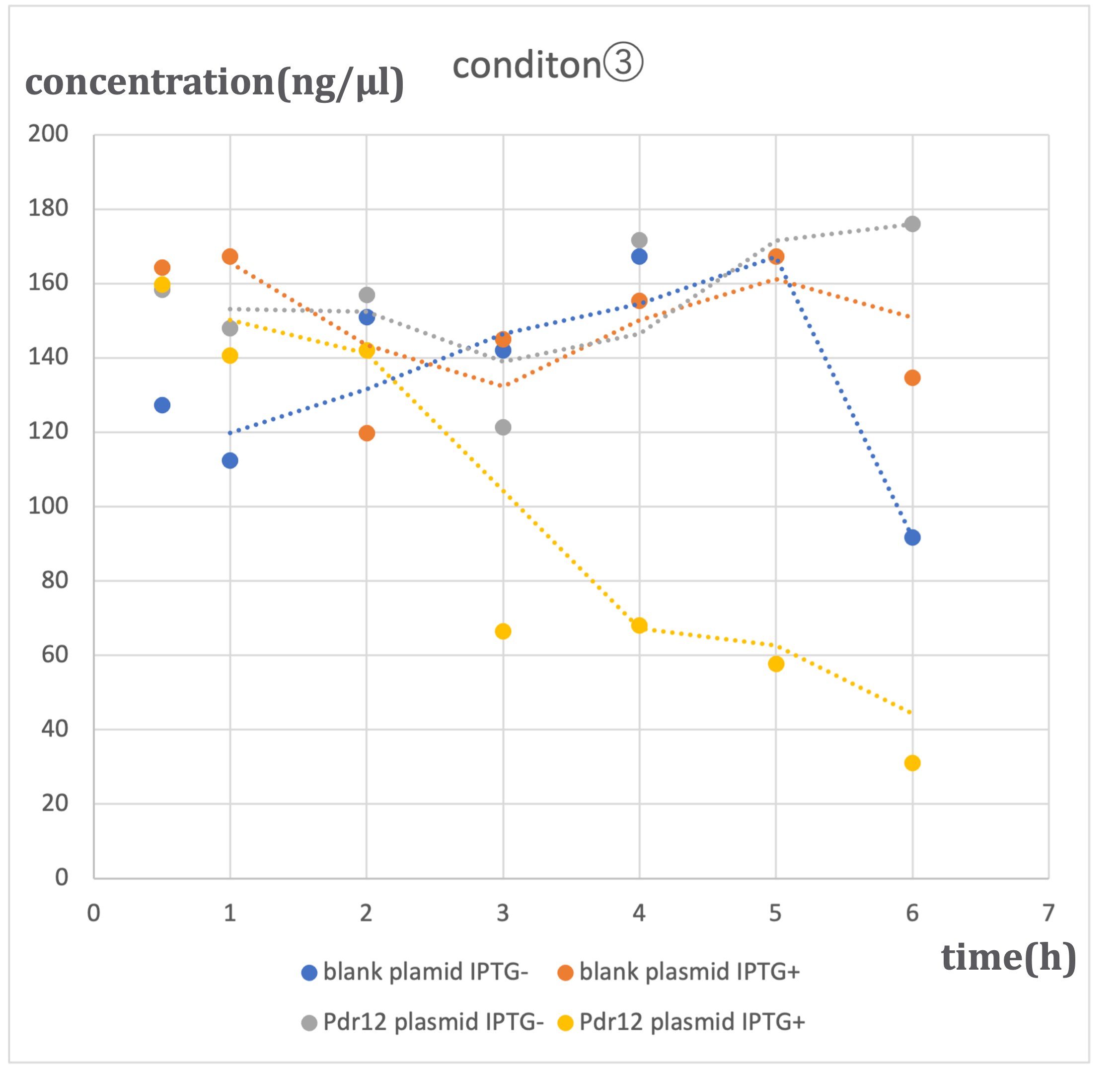
Learn
Similar trends were observed under three different alanine concentrations (500μM-①, 1mM-②, 2mM-③). In conditions where the blank plasmid was induced with IPTG (hereinafter referred to as blank plasmid IPTG+), as well as in conditions where it was not induced with IPTG (hereinafter referred to as blank plasmid IPTG-), and in conditions where the Pdr12 plasmid was not induced with IPTG (hereinafter referred to as Pdr12 plasmid IPTG-), a slight increase in pyruvate concentration was observed over time.
In contrast, under conditions where the Pdr12 plasmid was induced with IPTG (hereinafter referred to as Pdr12 plasmid IPTG+), a decrease in pyruvate concentration was observed at all alanine concentrations.
It was anticipated that pyruvate concentration would increase, especially under Pdr12 plasmid IPTG+ conditions. However, the actual results differed. The likely cause of this result is the increased expression of Pdr12 protein due to IPTG induction, resulting in an increased demand for pyruvate within Escherichia coli and the transportation of pyruvate into the bacterial cells through membrane proteins. Despite the knockout of the aceF gene, it was expected that pyruvate consumption would be somewhat inhibited, but the effect was limited, and the fundamental demand of E. coli remained unchanged.
The lack of a significant decrease in pyruvate concentration under blank plasmid IPTG+ conditions suggests that the expression of β-galactosidase induced by IPTG alone may not lead to a substantial change in pyruvate demand. On the other hand, the Pdr12 protein is expressed from a gene of approximately 4000bp, which suggests that it may have a significant impact on E. coli metabolism.
References
- Casal, Margarida, et al. "Transport of carboxylic acids in yeasts." FEMS microbiology reviews 32.6 (2008): 974-994.
- Hazelwood, Lucie A., et al. "A new physiological role for Pdr12p in Saccharomyces cerevisiae: export of aromatic and branched-chain organic acids produced in amino acid catabolism." FEMS yeast research 6.6 (2006): 937-945.
- Liu, Ke, et al. "Biotransformation and chiral resolution of d, l‐alanine into pyruvate and d‐alanine with a whole‐cell biocatalyst expressing l‐amino acid deaminase." Biotechnology and Applied Biochemistry 67.4 (2020): 668-676.
- Hossain, Gazi Sakir, et al. "Transporter engineering and enzyme evolution for pyruvate production from D/L-alanine with a whole-cell biocatalyst expressing L-amino acid deaminase from Proteus mirabilis." RSC advances 6.86 (2016): 82676-82684.
- Tomar, A., M. A. Eiteman, and E. Altman. "The effect of acetate pathway mutations on the production of pyruvate in Escherichia coli." Applied microbiology and biotechnology 62 (2003): 76-82.
- Dave, Ushmaben Chandrakantbhai, and Ravi-Kumar Kadeppagari. "Alanine dehydrogenase and its applications–A review." Critical reviews in biotechnology 39.5 (2019): 648-664.
- Vieira J and Messing J.Methods in Enzymology. (1987) 153: 3-11.
- Boliver F, Rodrigues R L, Green, P J, Betlach M C, Heynecker H L,Boyer H W, Grosa J H, and Falkow S.Gene. (1977) 2: 95-113.