Results
Quick Links
Choosing the appropriate vector
Prepare vector-insert construct
Expression and Purification
Transformation of E. coli auxotroph strain
Future Steps

Outline: Results and Discussion
Choosing the appropriate vector
In order to synthesize the bacterial collagen-like protein (Scl2.28), we decided to use the pQE80L-Kan plasmid from Qiagen. The plasmid places a 6XHis tag at the N-terminus of the protein of interest allowing subsequent purification using Ni-NTA affinity chromatography. pQE80L was specifically chosen as it encodes a cis-lacIq gene, which makes it not require the pREP4 repressor plasmid compared to other pQE vectors. While other vectors, such as the pColdIII has been extensively used in literature [1, 2], the use of pQE80L is recommended for fibril formation studies of bacterial collagen-like protein and the incorporation of ncAAs [3], in our case, hydroxyproline and hydroxylysine.
Ideally, we would choose pQE80L with resistance to ampicillin, however, the only vector we could import on time was pQE80L with Kanamycin resistance. This makes is slightly tricky as the E. coli Keio Knockout Collection [4] from which we obtained the proline and lysine auxotroph strains, JW0233 and JW2806 replaces the deleted proA and lysA gene with a kanamycin resistance cassette. FLP recognition target (FRT) recombination can be used to excise the kanamycin resistance; however, this is not something that we could try.
Prepare vector-insert construct
One of the challenges that we faced was the cloning of the gene of interest into the pQE80L vectors. Further details are documented on the Engineering Success page. We considered both Gibson Assembly and cloning using restriction enzymes. Part of the consideration was the practicality of the option as ordering of complete Scl2.28 was not possible through IDT due to high complexity. While we were able to amplify the fragments for Gibson Assembly, we could not obtain an assembled plasmid with the pQE80L and the fragments in appropriate order using NEB HiFI DNA Assembly. However, previous iGEM team (Collagene, 2021) has reported being able to assemble the plasmid using Gibson Assembly. It may be the case that the assembly conditions needs to be optimized.
For cloning using restriction digestion, we had to go through several iterations of ligating the Scl2.28 with various combinations of pQE80L-Kan. Specifically, we tried out the following combinations:
- pQE80L double digested for 15 mins ligated to double digested pUC57-Kan with Scl2.28 using T7 ligase.
- pQE80L double digested for 30 mins (A), 1 hour (B), and 2 hours (C) ligated to double digested pUC57-Kan with Scl2.28 using T7 ligase.
- pQE80L digested for 20 mins using BamHI followed by 20 mins using HindIII ligated with double digested pUC57-Kan with Scl2.28 using T4 ligase.
- pQE80L double digested for 1 hour ligated to double digested pUC57-Kan with Scl2.28 using T4 ligase.
Ligation reaction 1 and subsequent transformation in BL21 resulted in single colonies. To verify the construct, plasmid amplification and purification was performed. The purified plasmid was double digested. The bands obtained were the same as the bands for the pUC57-Kan with Scl2.28. This indicated to us that the digestion may not be going to completion and residual pUC57-Kan could interfere with subsequent screening. Hence, the digestion time was increased to 30 minutes to an hour for Scl2.28.
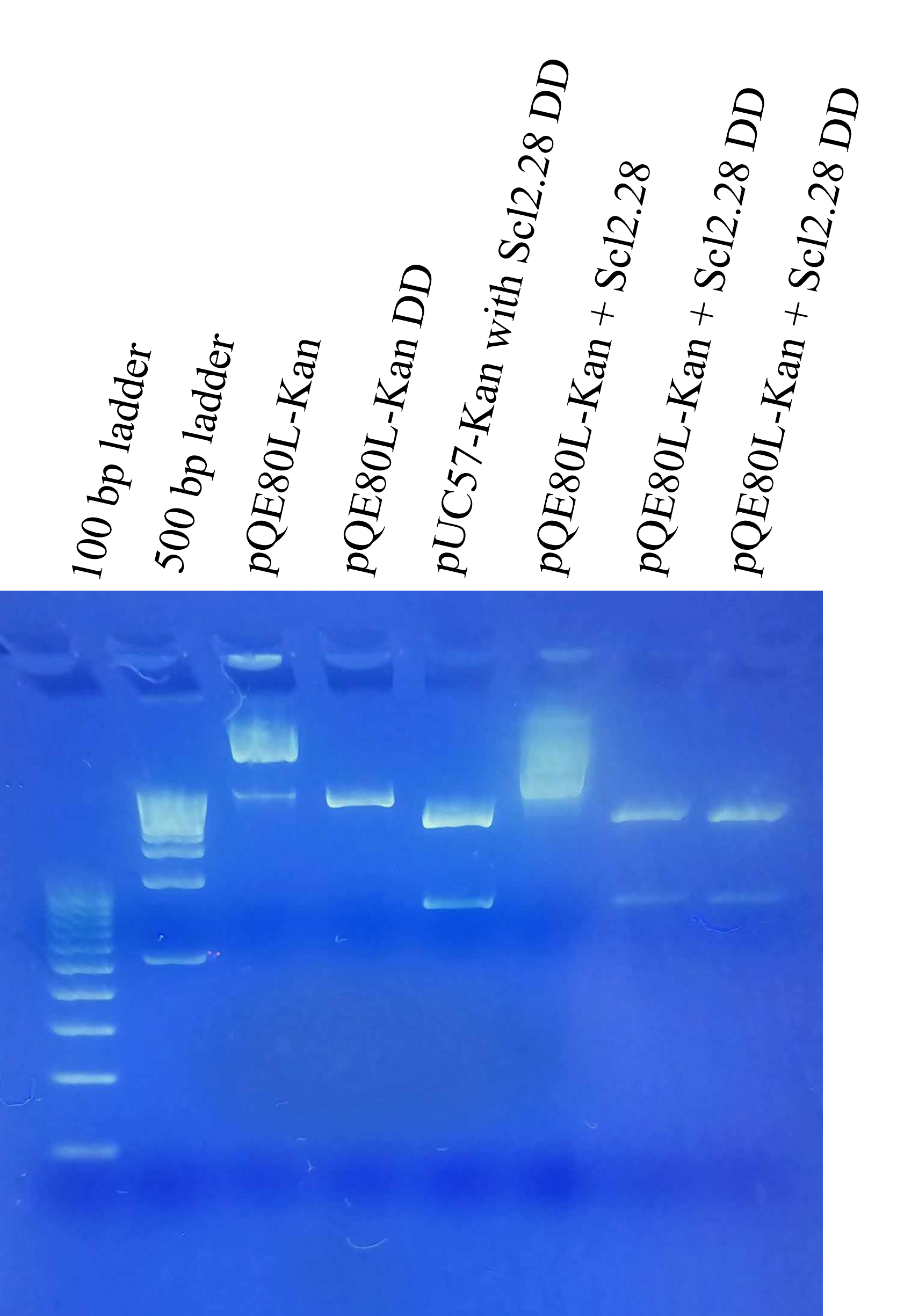
Following this, we transformed the plasmids obtained in combinations 2A, 2B, 2C, 3, and 4. Subsequently, the plasmids were amplified and double digested to check for desired band in gel electrophoresis as shown below. From these gels, we shortlisted two possible candidates for further characterization, namely 2C and 4, as they had the bands that matched double digested pQE80L-Kan (4500 bp) and the insert sequence of pUC57-Kan with Scl2.28 (711 bp). We theorized two possible scenarios for 4, one that the ligation was unsuccessful because of failed digestion and both pQE80L-Kan and pUC57-Kan were present. The second scenario was that ligation was successful, but excess pUC57-Kan was still present. The wide bands of the undigested ligated product also made it difficult to estimate the size of the ligated plasmids. To get definitive verification of ligation, primers were designed and ordered for PCR. The forward primer was on the insert sequence, while the reverse primer was on the vector backbone.

For both the constructs (2C and 4), PCR product was visible. The PCR band was wide but fell in the appropriate size (~800 bp). No PCR product was visible for negative control. This indicated successful generation of pQE80L-Kan+Scl2.28 plasmid. These plasmids were then used to transform BL21 and used for subsequent expression.

Expression and Purification
Overnight liquid culture of both 2C and 4 was added to 150 mL LB Broth and expression was induced using IPTG once the OD600 reached 0.5. For each construct, one sample was allowed to incubate for 6 hours and one for 16 hours. At the end of each duration, cells were pelleted. Cell pellets were used for purification following Qiagen Ni-NTA Fast Start Kit protocols due to easy of preparing buffer and sample volume. The non-induced control, induced control, cell lysate, flow through, wash fraction, and elution fractions were analyzed using SDS-PAGE followed by Coomassie stain. A distinct band approximately 35 kDa corresponding to Scl2.28 is visible across all the SDS-PAGE in the induced controls. This value is in close agreement with value from literature [3]. However, the purification protocol needs to be optimized. For example, the Scl2.28 protein band is apparent in lane 7 of SDS-PAGE of ligation combination 4 (6 hours expression) corresponding to the washing step of the purification. This could be due to the buffer composition of the kit. The specific components of the buffer are not available in the kit handbook.


Transformation of E. coli auxotroph strain
E. coli JW0233 and JW2806 auxotroph strains were made competent using calcium chloride method. Competent cells were then transformed with plasmid 2C and 4 and plated on kanamycin and chloramphenicol agar plates. pUC19 was used as a positive control and plated on ampicillin agar plates. Growth was present in positive control plates indicating auxotroph strains were competent. Single colonies were not visible in Kan plates but was present in Cam plates.

Future Steps
Purification needs to be repeated with revised buffer composition based on protocol available in literature [3] and expert consultation. Protocol is available on the experiments page. Once the protocol has been optimized, protein can be purified in larger quantities.
For residue-specific incorporation of hydroxyproline and hydroxylysine, growth curve optimization of auxotroph strains in M9 media containing various concentrations of proline and lysine respectively needs to be performed. From the growth curve analysis, the concentration at which the cells reach a plateau needs to be determined and indicates proline depletion. Subsequently, larger scale liquid cultures in M9 media can be cultured at the optimal concentration of proline or lysine and once culture reaches the desired OD, expression is induced using IPTG alongside the addition of hydroxyproline and hydroxylysine. We intend to compare two methods for residue-specific incorporation of Hyp ang Hyl, proline depletion as outlines here, and media shift as outlined by Breunig [5].
After producing the modified CLP, we aim to characterize its properties. Fourier-transform infrared spectroscopy (FTIR) will be used to analyze morphology and secondary structure, circular dichroism to investigate the triple-helical structure, and dynamic light scattering to assess self-assembly capabilities. We will also incorporate cell-adhesion domains through integrin binding sites and other functional domains to enhance interaction with cells. Cytotoxicity and biocompatibility tests will be performed to ensure safety for tissue engineering applications. We hope to explore the CLP's hydrogelation capabilities for 3D bioprinting through the addition of methacrylate groups for photo crosslinking or 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) chemical crosslinking.
The ability to design collagen-like protein is a salient feature made possible through synthetic biology. Tailoring the properties of collagen-like protein, like Scl2.28, has huge potential for enhancing tunability and improving the level of control over systems.
References
- Mohs, A., et al., Mechanism of Stabilization of a Bacterial Collagen Triple Helix in the Absence of Hydroxyproline*. Journal of Biological Chemistry, 2007. 282(41): p. 29757-29765.
- Yoshizumi, A., et al., Self‐association of Streptococcus pyogenes collagen‐like constructs into higher order structures. Protein Science, 2009. 18(6): p. 1241-1251.
- Ilamaran, M., et al., A self-assembly and higher order structure forming triple helical protein as a novel biomaterial for cell proliferation. Biomaterials Science, 2019. 7(5): p. 2191-2199.
- Baba, T., et al., Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular Systems Biology, 2006. 2(1): p. 2006.0008.
- Breunig, S.L. and D.A. Tirrell, Chapter Seventeen - Incorporation of proline analogs into recombinant proteins expressed in Escherichia coli, in Methods in Enzymology, E.J. Petersson, Editor. 2021, Academic Press. p. 545-571.